

Articles sur le même thème :


Proteoliposomes – ideal model systems to study the structure and function of membrane proteins in semi-native environment by cryogenic electron microscopy
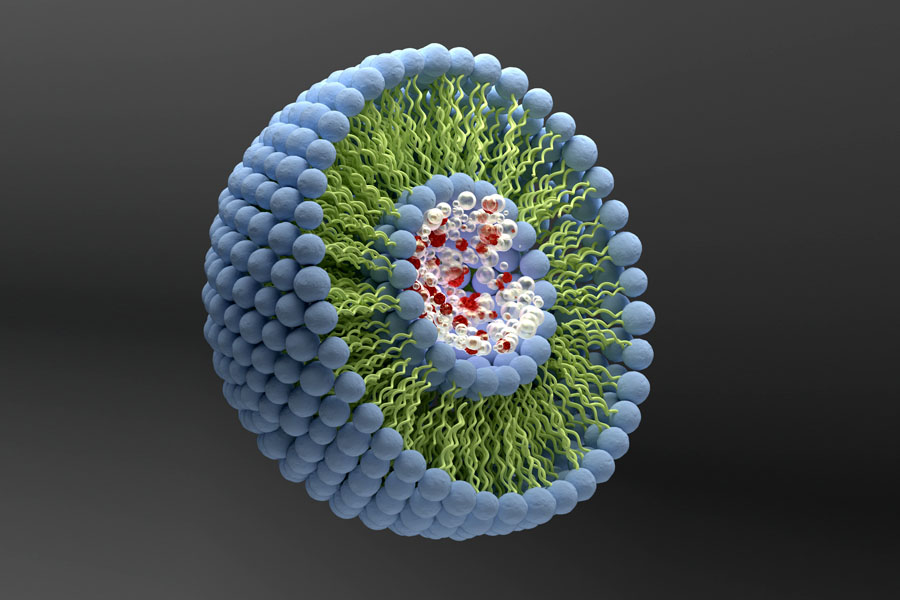
What are Proteoliposomes ?
Proteoliposomes are lipid vesicles containing membrane proteins that can be used as model systems to perform structural and functional studies of membrane proteins under conditions that partially mimic their native environment.
Here we present the conventional methods for liposome formation, the strategies to insert purified membrane proteins into the bilayer of unilamellar liposomes and the use of cryogenic electron microscopy to perform structural analysis of liposomes and proteoliposomes. In particular, we refer to the approach of using proteoliposomes and cryogenic electron microscopy to capture pictures of the membrane proteins in action under semi-natural conditions.
Liposomes are microscopic vesicles formed by one or more concentric amphiphilic lipidic bilayers separated by aqueous medium. They can encapsulate hydrophilic or lipophilic substances (in the aqueous compartment or inserted on the bilayer, respectively), are biocompatible and nonimmunogenic and, therefore, are used in therapeutic applications, as nanocarriers for drug delivery [1].
They can also be used as nanocarriers for functional proteins, for detoxification of drugs and endogenous metabolites, and as nanocompartments for crystal growth. Furthermore, they can be employed in the biosensing field, in nanosensors for biological targeting [2]. Proteoliposomes are a particular type of liposomes in which proteins are inserted in the lipid bilayer. Due to their composition, proteoliposomes can be used as model systems to study biological membranes and their proteins.
Why are they good model systems to study membrane proteins ?
The grounds to analyze membrane proteins rely on their important roles in biological processes, such as mediation of energy transport during photosynthesis or respiration, molecule transport across the membrane, transmission of chemical signals, cytoskeleton anchoring and catalysis of chemical reactions. The numerous functions of membrane proteins justify the fact that they are the target of the majority of available drugs [3]. Nevertheless, structural studies are still difficult to perform and new methodologies to analyze membrane proteins are needed.
Proteoliposomes are, thus, a promising and advantageous approach, as they enable to characterize membrane proteins in the presence of a membrane potential or ligand gradient [4]. Moreover, it has become clear that the structure of membrane proteins should be studied in close-to-native conditions, as the differences in transmembrane potential cause conformational changes of membrane proteins and, consequently, affect their functions [5]. The possibility offered by proteoliposomes to study membrane proteins under semi-native conditions reinforces their potential as model systems.
The different ways to prepare them
Usually, liposomes can be obtained by the film dispersion method or by the dialysis method. In the first method, lipids are solubilized with chloroform, followed by solvent evaporation, which results in the formation of a lipid film on the recipient walls. The lipid film is then dispersed in the chosen aqueous buffer or buffer plus additives (for encapsulation experiments) by vortexing. When working with phospholipids, different types of vesicles can be obtained, namely multilamellar vesicles and large unilamellar vesicles. A further step of extrusion results in the formation of small unilamellar vesicles. A final size exclusion chromatography procedure can be performed to obtain more homogeneous size distributions [6].
In the dialysis method, the lipid film is solubilized in a buffer containing detergent in a concentration above its critical micellar concentration (cmc) and loaded into dialysis buttons or a dialysis bag, where it is dialyzed against detergent-free buffer for hours to weeks. As the detergent concentration decreases below cmc, liposomes are formed. Finally, the liposome mixture must be extruded through a filter several times to obtain homogeneous unilamellar vesicles. A last size exclusion chromatography step can also be carried out to decrease the size distribution or to exchange the dispersion buffer for encapsulation experiments [6,7].
Reconstitution of membrane proteins into liposomes is usually performed by mixing detergent-solubilized purified membrane proteins in buffer solution with detergent-solubilized lipids, followed by slow reduction of detergent amount by dialysis [7], by the addition of cyclodextrin [8] or by gradual dilution [7]. Several experimental conditions, such as detergent, buffer, pH, additional salts, lipid-to-protein ratio, temperature gradients and time scale must be optimized to generate the structure of interest [7].
Another method for protein reconstitution in liposomes is based on cell-free protein synthesis (CFPS). This method uses the transcription and translation machinery of cells to express membrane proteins in vitro [9]. By mixing them with preformed liposomes, membrane proteins are directly inserted into liposomes during production.
What can you get from their analysis ?
Electron microscopy, and particularly cryogenic electron microscopy (cryo-EM), is frequently used to analyze liposomes and proteoliposomes. To perform cryo-EM, the liposome or proteoliposome liquid samples are applied onto the cryo-EM grid, which is then blotted with filter paper and rapidly frozen, and the imaging is performed at cryogenic temperatures [10]. The micrographs are recorded at a range of defocus settings under low electron-dose conditions to minimize beam damage.
Cryo-EM provides information about sample homogeneity and vesicle morphology, enables to measure membrane thickness, exposes the location of the proteins and their structure, and allows to observe membrane protein crystals. Furthermore, when combined with electron tomography, it reveals the 3D shape of liposomes in solution. Cryo-EM also enables to see the effect of additives, encapsulation and protein-liposome interactions [11].
Notwithstanding, some misleading artifacts can occur during sample application, confounding the imaging of liposomes and proteoliposomes. These artifacts are caused by variations in the thickness of the vitrified sample layer and by drying of the sample solution with the consequent increase in salt concentration and vesicle deformation or breaking. In addition, because almost of the liquid deposited on the grid is removed by paper blotting, only a qualitative analysis can be performed. The absolute determination of the mean liposome diameter or the relative occurrence of different liposome species is not possible [6].
Membrane protein in action !
Apart from all the challenges that still remain in the analysis of membrane proteins, the use of proteoliposomes combined with cryo-EM has open the possibility to characterize membrane proteins in action. To achieve this, a new strategy was proposed, in which unilamellar liposomes are firstly formed in buffer A, which encapsulate that buffer in their lumen, and reconstitute the membrane protein into the liposome bilayer using the same buffer.
The outer buffer of this proteoliposome suspension can be exchanged to buffer B and the structure of the reconstituted membrane protein can be imaged by cryo-EM and electron crystallography. After buffer exchange, buffer A must remain encapsulated for some time, during which the environment is different on both sides of the bilayer and sample grids can be rapidly frozen for cryo-EM, allowing to image a picture of the membrane protein in action and to observe the conformation changes involved.
In the future, this strategy could allow to study structure-function relations of membrane proteins by generating buffer or ligand gradients across the lipid bilayer of proteoliposomes [6].
Synthelis
As a service provider with more than 10 years of experience in cell-free technology and membrane protein expression, Synthelis is able to produce membrane proteins in proteoliposome format using either its patented approach to directly incorporate the protein target into the lipid bilayer of the liposomes, or after protein solubilization and purification followed by a relipidation phase. Both approaches have their pros and cons.
Our team has also the know-how and the experience to produce membrane proteins either solubilized in detergent or in nanodisc format.
If you have such project and want to discuss its feasibility, please contact us.
Sources
[1] Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 6 (2015) 286.
[2] Chen C, Wang Q. Liposome-based nanosensors for biological detection. Am. J. Nano Res. Appl. 3 (2015) 13-17.
[3] Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Computational analysis of membrane proteins: the largest class of drug targets. Drug Discov. Today 14 (2009) 1130-1135.
[4] Banerjee RK, Datta AG. Proteoliposome as the model for the study of membrane- bound enzymes and transport proteins. Mol. Cell Biochem. 50 (1983) 3-15.
[5] Bezanilla F. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 9 (2008) 323-332.
[6] Sejwal K, Chami M, Baumgartner P, Kowal J, Müller SA, Stahlberg H. Proteoliposomes – a system to study membrane proteins under buffer gradients by cryo-EM. Nanotechnol. Rev. 6 (2017) 57-74.
[7] Rigaud JL, Lévy D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 372 (2003) 65-86.
[8] Signorell GA, Kaufmann TC, Kukulski W, Engel A, Remigy HW. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J. Struct. Biol. 157 (2007) 321-328.
[9] Whittaker JW. Cell-free protein synthesis: the state of the art. Biotechnol. Lett. 35 (2013) 143-152.
[10] Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21 (1988) 129-228.
[11] Almgren M, Edwards K, Karlsson G. Cryo transmission electron microscopy of liposomes and related structures. Colloids and Surfaces A 174 (2000) 3-21.